


Estimating Mutational Pressure on IQ through the Effect of De Novo Mutations in the Exome on Intellectual Disability
Molecular data shows the mutational pressure on IQ is significant

This article was motivated by the following X-change:

This is pretty cool data. The abstract:
There are established associations between advanced paternal age and offspring risk for psychiatric and developmental disorders. These are commonly attributed to genetic mutations, especially de novo single nucleotide variants (dnSNVs), that accumulate with increasing paternal age. However, the actual magnitude of risk from such mutations in the male germline is unknown. Quantifying this risk would clarify the clinical significance of delayed paternity. Using parent-child trio whole-exome-sequencing data, we estimate the relationship between paternal-age-related dnSNVs and risk for five disorders: autism spectrum disorder (ASD), congenital heart disease, neurodevelopmental disorders with epilepsy, intellectual disability and schizophrenia (SCZ). Using Danish registry data, we investigate whether epidemiologic associations between each disorder and older fatherhood are consistent with the estimated role of dnSNVs. We find that paternal-age-related dnSNVs confer a small amount of risk for these disorders. For ASD and SCZ, epidemiologic associations with delayed paternity reflect factors that may not increase with age.
Are these estimates causal? Apparently yes, they have genotype-level data on how many dnSNVs (a subset of de novo mutations) the kids have. They find the effect of measured dnSNVs and then adjust this by the rate at which fathers accumulate dnSNVs in the sperm.
Its development is described in detail in the Methods. The output of the model is an incidence rate ratio (IRR) reflecting the increase in dnSNV-related disease risk in offspring of older fathers compared with offspring of younger fathers. In brief, one can estimate disease incidence in offspring of fathers of a specified age (e.g. people born when their fathers were 25) by multiplying (a) disease incidence among individuals who carry zero dnSNVs (INCbaseline) and (b) the odds ratio (OR) associated with one additional nonsynonymous dnSNV, exponentiated to the expected number of dnSNVs in offspring of fathers of the specified age (see Methods, below). Baseline incidence can be unknown or variable with respect to time as, algebraically, it is not needed for estimating the IRR. As noted in Equation (1), effect size variation across different types of dnSNVs is also algebraically irrelevant under the assumption that all types of dnSNVs increase with age at the same rate (Supplementary Notes 1 and 2).
To show this we use the example of missense and protein truncating variants (PTVs), which have different average effect sizes on risk for ASD. This logic also applies to post-zygotic mutations (PZM) that are confused for de novo variants. PZMs would not affect our estimates under the assumption that PZM rate is unassociated with paternal age (Supplementary Note 3).
As described in Supplementary Notes 4 and 5 and Supplementary Fig. 1, the model’s estimates are also robust to: plausible variation in the expected (control) rate of dnSNVs, plausible variation in the estimated effect size of case dnSNVs, and the inclusion of de novo copy number variants. The model assumes that risk-conferring statistical interaction among dnSNVs do not commonly occur and additionally assumes, contrary to the selfish spermatogonial selection model37, that de novo variants that emerge in sperm cells later in life are no more (or less) likely to be disease-associated than other de novo variants. In Supplementary Note 6 we show that interactions among disease-associated dnSNVs are not currently identifiable in the ASD data and discuss why they are unlikely to be relevant for any of the other disorders. In supplementary Note 7 we discuss the selfish spermatogonial selection theory, discuss the implications for our model if such a mechanism were significant, and explain why this phenomenon is unlikely to substantially influence our findings.
Because not every cohort used the same technology for sequencing individuals or the same procedure for calling dnSNVs, we adjusted dnSNV rate in each cohort by its rate of synonymous de novo variation (See Methods, below). We did so based on the assumption that the true rate of synonymous variation across cohorts is approximately equal. In Supplementary Note 8 we show that the results we describe here are robust to whether or not one accepts this assumption (Supplementary Figs 2 and 3; Supplementary Tables 1 and 2). Finally, in Supplementary Note 9 we show that the results are not substantially affected by modeling dnSNV accumulation to begin at puberty (approximated by age 13).
The key result is here:

These seem to be a bit less than estimates for the same traits coming from the best non-molecular method, which is within-families analysis of the effect of paternal age on a trait.

IRR from the first study seems to just be the OR through a dnSNV multiplied by itself the number of times equal to the number of dnSNV present on average at different paternal ages, whereas the OR from the latter study is the increase in incidence for paternal age groupings relative to the 25-29 year old group, controlled for basically everything. The sibling-comparison data is not confounded for older fathers or mothers being more likely to have higher or lower gene scores, birth order, or maternal effects. It should reflect a robust estimate of the effect of paternal age on a trait, but this may not be all dnSNV . It could also involve mutations to areas they did not sequence. On this the authors comment thus:
While we have explicitly considered the role of de novo mutations in the exome, it is possible that de novo single nucleotide variants in the rest of the genome could create additional risk associated with advanced paternal age. However, there is currently no evidence of which we are aware for enrichment of de novo variants outside of the exome in genetically complex human diseases35. More importantly, if de novo variants that do not effect protein structure did contribute sufficiently to some genetically complex disorders (ASD, SCZ) to cause a large increase in their epidemiologic association with advanced paternal age, it would be difficult to account biologically for the fact that the mutational burden in the exome explains the association between advanced paternal age other genetically complex disorders (CHD, EPI and ID) as we show here.
The exome is apparently only 1.5% of the genome. I think it’s pretty likely that congenital mutations in the other 98.5% can create some issues.

As for the paternal age relationship for some diseases being accounted for by this data, not really. This mostly comes out of noise in the epidemiological data they use. Considering the the sibling comparison data above, it’s clear their estimates, taken as a group, are consistently lower than the epidemiological estimates except in the case of heart disease, where the epidemiological data is sparser and noisier.
In addition to this data not accounting for the effect of mutations outside the exome, it would also miss the effect of an increase in paternal age on a child that operates through older fathers parenting differently. While mutations outside the exome would accumulate across generations, such a “cultural” effect would not. However, it’s unlikely such an effect is significant for highly heritable traits like IQ and mental retardation.
Estimating the Mutational Pressure with this Data
With one assumption, we can compute the number of IQ points lost per generation due to exome dSNVs using this data. That assumption is that intellectual disability (ID) is a thresholding function of IQ. That is, if 2% of people have ID, they are simply the bottom 2% of people by IQ.

This seems to indeed be the definition used by clinicians. It seems like ID is diagnosed by scoring under 2 SDs on a population-normed, normally distributed IQ test.
We can use the following math. First, recall the equation for mutational pressure:

It’s about 3 times the robust correlation between paternal age and a trait, because that correlation is how many SDs a trait decreases by per SD of paternal age (7 years), and each generation has fathers that have accumulated about 21 years worth of mutations, on average.
If we look again at the main results from the study, we see that 20 years of aging increases the rate of ID by 1.1 to 1.5 times.
The SD change from a paternal age of 25 to 45 is just
Where phi^-1 is just the inverse CDF of the standard normal.

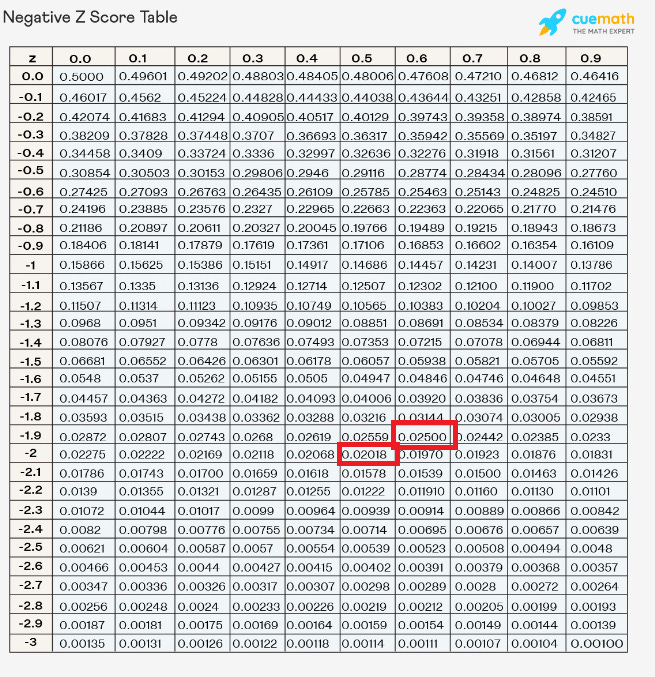
So if it’s 2% at paternal age 25, and it becomes 2.5% at paternal age of 45, then, with the mean held constant, those with ID have a z score of -2.05 at a paternal age of 25 and a z score of -1.96 at the paternal age of 45. If ID is defined as everyone in the bottom 2% of the PA25 distribution, then the mean IQ of the PA45 distribution must be 0.054 SDs less than the PA25 distribution mean, in order for the bottom 2.5% of the PA45 distribution to be equally as retarded as the bottom 2% of the PA25 distribution.
This is 20/7 = 2.85 paternal age SDs, so the effect of 1 paternal age SD should be -.09 / 2.85 = -0.03. This should be r_a,g because it’s the effect of 1 SD of paternal age on the trait in terms of SDs. The mutational pressure is then -.09 = -1.35 points per generation.
This is in the 95% CI for the paper I’ve previously gone off of for mutational load and IQ. This paper controlled for paternal IQ polygenic score, but was epidemiological in that it did not measure any mutations, it only observed the effect of paternal age on IQ with some controls. Further, it lacked the power to properly control for birth order effects.
Bayesian analysis
Let’s make the incidence rate of ID and the IRR random variables, because there’s some uncertainty over exactly what they are.
import numpy as np
from scipy.stats import norm
import matplotlib.pyplot as plt
def phi_inv(percentile):
return norm.ppf(percentile)
id_or = np.random.normal(1.3, .1, 10000000)
id_rate = np.random.normal(0.02, .004, 10000000)
new_id_rate = (id_rate * id_or)
sd_change = phi_inv(id_rate) - phi_inv(new_id_rate)
sd_change_per_pa_sd = sd_change / (20/7)
plt.figure(figsize=(10, 6))
plt.hist(sd_change_per_pa_sd, bins=50, color='skyblue', edgecolor='black')
plt.title('Distribution of Standard Deviation Change per Paternal Age SD (sd_change_per_pa_sd)')
plt.xlabel('IQ SD Change per 7 Paternal Age Years from exome dnSNVs ')
plt.ylabel('Frequency')
plt.grid(True)
plt.show()
IQ_mutational_pressure = sd_change_per_pa_sd * 3 * 15
plt.figure(figsize=(10, 6))
plt.hist(IQ_mutational_pressure, bins=50, color='skyblue', edgecolor='black')
plt.title('Distribution of Mutational Pressure on IQ per Generation (IQ scale) (IQ_mutational_pressure)')
plt.xlabel('Mutational Pressure on IQ per Generation (IQ scale) from exome dnSNVs ')
plt.ylabel('Frequency')
plt.grid(True)
plt.show()
We get the above output.
Considering that this is only from exome dnSNVs and not other mutations, I’d say this is good evidence that the mutational pressure on IQ is between -1 and -3 points per generation, if not more. There’s still a small chance from this data that it’s little to nothing, but it’s unlikely from this data alone. Considering all of the non-molecular data and other robustly demonstrated paternal age effects (e.g. from within family studies), as well as the way in which this method gives a lower bound, it should be less likely than the chart indicates for the mutational pressure on IQ to be lower magnitude than -1 point per generation.
Circling back the the X-change that motivated this, my estimate was potentially off by a factor of 2, which is because the Wang paper gave a point estimate of -5 points per generation. However, it’s possible that non-exome mutations cause 1 or 2 point loss every generation, but there’s no way of knowing right now, to my knowledge.
I can amend my statement: Every 7 years of paternal age beyond 14 knocks about 0.05 SD off of expected value of child IQ due to exome mutations. So if you are 49, that's -0.25 SD from your gene scores. If you are 2 SD, you average kid could still be 1.75 SD, so it could still improve the gene pool to breed. This may not account for mutations to the non-coding region, but those almost certainly drop less than another 0.05 SD off per 7 years of paternal age, and not more.
I wonder how much better my substacks would be if my father was 10 years younger when he had me… Also, thoughts on the theory that paternal age is a factor in national IQ score differences? Some people suggest that MENA/Africa are low IQ because of higher paternal ages. You know, old village lords having their young wives. However, all the data I have seen suggests that people have kids younger in non western countries.